
Prehistoric Peopling In Southeast Asia: Genomics Of Jomon And Other Ancient Skeletons
Uncovering the expansion processes of human habitats in the past is of great importance for understanding the origins and establishment of present-day populations and the acquisition of genetic characteristics of individuals as well as for investigating mechanisms of resistance against diseases and pathogens.
Previous genetic/genomic studies aimed to uncover the expansion processes using present-day human genomes of different individuals and locations. However, it is not always possible to elucidate the expansion processes based on the genomic similarity of present-day populations due to the possibility of migrations of populations between regions in various periods. It is therefore impossible to uncover the precise expansion processes of populations in the past without knowledge of the genomic information existing in a designated region and period. Thus, expansion processes hypothesized so far were nothing but speculations based on assumptions about present-day genomes.
Recent developments of DNA analysis technology have made it possible to obtain whole genome information from ultratrace amounts of DNA; we are now in an era where whole genome information can be obtained directly from ancient human skeletons discovered at archaeological sites. There remain, however, technical problems for obtaining whole genome information of ancient human skeletons. In particular, there are two main problems: first, genomic analyses*1) of poorly-preserved ancient remains in hot and humid regions of the world have up until now failed.
Secondly, there is the risk of contamination of present-day human DNA in the DNA samples of ultratrace amounts from prehistoric remains. To evaluate objectively the possibility of such contamination, several different research groups must cross-check*2) one another in order to achieve exact genome sequencing; in other words, establishment of a collaborative research system is a prerequisite for attaining the highest level of scientific authenticity.
In order to cope with these problems, the present international research team, led by researchers from the University of Copenhagen with the participation of three researchers from Kanazawa University has established technologies to efficiently extract human DNA from skeletons discovered at prehistoric remains even under very poor conditions for DNA preservation. At the same time, an international system of research collaboration has been established for objectively evaluating the effects of contamination by present-day human DNA. Thanks to these efforts, the team has uncovered the expansion processes of human habitats and genetic interactions in hot and wet Southeast Asia, which was not possible previously with conventional technologies and research systems.
Worthy of special mention, the present study has been successful in determining the “whole genome” sequence of an individual with typical Jomon culture, while previous studies were only able to show a very limited “partial genome” sequence of two Jomon individuals. Thus, the present study is the first successful example to show the possibility of whole genome sequencing of prehistoric individuals in regions like Japan where preservation conditions are quite poor, possibly leading to further major progress in prehistoric genome studies.
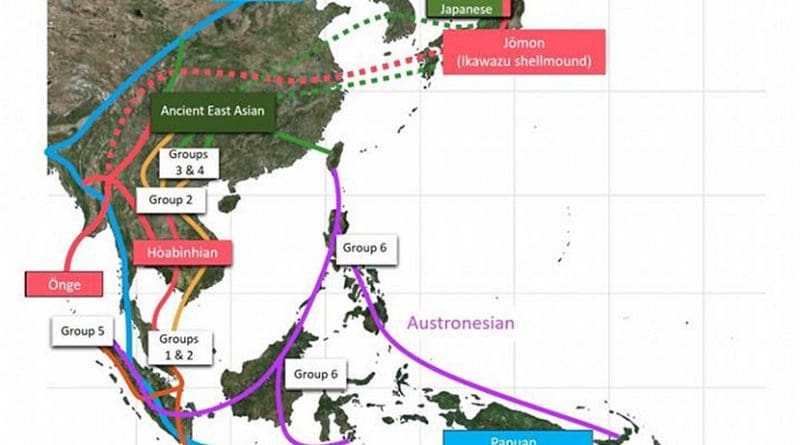
In the present study, the international research team succeeded in extracting and sequencing DNA from 25 ancient individuals’ skeletons from Southeast Asian remains, where the condition of DNA preservation is very poor, and from one Japanese Jomon female skeleton. Upon comparison of the genomic data of ancient human skeletons with those of present-day human skeletons, it has become clear that those prehistoric populations in Southeast Asia can be classified into six groups.
Group 1 contains Hoabinhians from Pha Faen, Laos, hunter-gatherers (~8000 years ago), and prehistoric populations discovered from Gua Cha, Malaysia (~4000 years ago), being genetically close to present-day Önge and Jarawa from the Andaman Islands and Jehai from the Peninsular Malaysia. To our surprise, group 1 has higher genetic affinities with Ikawazu*3) Jomon individual (Tahara, Aichi), a female adult*4), than other present-day Southeast Asians. In addition, the Ikawazu Jomon genome*5) is best modelled contributing genetically present-day Japanese.
On the other hand, Groups 2-6 consist of ancient skeletons from the Neolithic Age, when farming started, until ~500 years ago. It is now found that they are genetically much different from Hoabinhians, each group having histories of migration and genetic interaction, i.e., inter-population mixture. Group 2 is found to be genetically close to the present-day Austroasiatic language-speaking groups such as Mlabri, but to have few genetic components common with the present-day East Asian populations. Group 3 is found to be genetically close to Kradai, Thailand, in the present-day Southeast Asian populations and to the Austronesian language-speaking groups. Group 4 is found to be genetically close to the present-day populations in South China. Group 5 is genetically close to the present-day populations in the western part of Indonesia. Group 6 is most closely related to present day Austronesian populations, with one individual showing slightly elevated Denisovan ancestry, an archaic hominin which is classified as a sister group of Neanderthals.
As above, Neolithic Southeast Asians are found to have been partially genetically influenced by ethnic groups in South China and to have had a genetic connection with populations in Taiwan; Neolithic Southeast Asians are found not to have been indigenous hunter-gatherers passively accepting farming but to have accepted farming gradually in the process of migrations of populations between the continent and islands. Conventional archaeology proposed the two-layer hypothesis that, in those periods, a large population with farming culture with rice and millet migrated into Southeast Asia and that they replaced the indigenous population. Additionally, the present study indicates that the genetic influence from South China with rice farming was only partial and that the migrating population did not replace the indigenous population completely. The present analysis shows that there were at least four big migration waves; migrations of Southeast Asians should be investigated with a new “complex model” framework.
The present study successfully elucidates for the first time the expansion/migration of prehistoric populations by genome analysis of skeletons discovered in Southeast Asia; conventionally, it was thought that such population expansion/migration could only be investigated using archaeological artifacts. An important outcome of the present study is that the same or analogous analyses could be applied to various regions to evaluate the history of population expansion/migration in much more detail and in a more scientific manner.
The genomic data obtained from ancient skeletons in Southeast Asia and from a Ikawazu Jomon individual provides an important basis for investigations on the origins of populations in wider East Asia. The whole genome information of a Jomon individual will be useful for direct comparison of genomic similarity with ancient East Asians of the corresponding period to Jomon in present-day Korea, China, Russia and others in the vicinity of the Japanese archipelago. More comparative studies are in progress on populations in wider areas.
Note that the whole genome sequence obtained in this study for a Jomon individual corresponds to the Draft Genome Sequence in the Human Genome Project for the present-day humans. We aim at Complete Genome Sequence with higher accuracy.
This study is an interdisciplinary undertaking combining anthropology and archaeology in a close collaboration, allowing us to establish ourselves at the starting point for research on the origin of Jomon and its diversity. By more genome analyses of more Jomon skeletons from different Jomon sites, genetic diversity of Jomon populations will be explored over the Japanese archipelago. It is expected through such studies that various interactions among Jomon groups should be revealed together with migrations of archaeological artifacts such as potteries and stone tools as well as migrations of populations. Based on the outcome of the present study, novel anthropological and archaeological approaches would be further developed.
Glossary:
*1) Genomic Analysis
Analysis of whole genome of a species.
*2) Cross-check analysis
Cross-check analysis is an analytical method that evaluates whether the same result will be produced irrespective of different research institutions, different analytical methods, and so on. It is an important scientific index in research on ancient DNA.
*3) Ikawazu kaizuka (shellmound)
A kaizuka (shellmound) site at Tahara city, Aichi prefecture, dating back to late and final Jomon period. One of the best known archaeological site of Jomon period, where more than 200 individual skeletons have been discovered from Meiji era till today. A number of renowned anthropologists like Profs. Yoshikiyo KOGANEI and Hisashi SUZUKI performed morphological research on prehistoric skeletons from this site. There are also other kaizuka sites in Tahara city, such as Yoshigo kaizuka and Hobi kaizuka, representative Jomon sites. Those sites have been well studied and many skeletons have been excavated.
*4) A female skeleton dating back to late Jomon period, ~2500 years ago.
A Jomon skeleton discovered from Ikawazu kaizuka site in 2010. Recent studies indicate the beginning of Yayoi period to be ~3000 years ago, but the arrival of Yayoi culture differed depending on regions. The female adult skeleton from Ikawazu kaizuka site is accompanied with a pottery that is validated to date back to the period of Gokanmori type pottery, indicating that the period was still Jomon at those sites in Atsumi peninsula, Aichi prefecture. In addition, the female skeleton analyzed here shows typical Jomon morphology.
*5) Whole genome sequence
Whole genome sequence is the total DNA sequence of a species covering not only DNA sequences for genes but also DNA sequences for non-gene regions.